

Supplier:
Bioworld Technology, Inc.Cat no: BS4685P
p-FANCA (S1149) Peptide
Prices direct from Bioworld Technology, Inc.
Quick response times
Exclusive Biosave savings/discounts
SPECIFICATIONS
Catalog Number
BS4685P
Applications
BNI
Form
1 mg/ml in DI water.
Purity
Synthetic peptide p-FANCA (S1149). (Note: the amino acid sequence is proprietary). The purity is > 98%.
Swiss Prot
O15360
Storage Temp
Store at 4 degree C short term. Aliquot and store at -20 degree C long term. Avoid freeze-thaw cycles.
Additional Info
This peptide can be used with studies using BS4685 p-FANCA (S1149) pAb.
Alternative Names
Fanconi anemia group A protein; Protein FACA; FAA; FACA; FANCH
SUPPLIER INFO









Applications
BNI
Applications
BNI
Applications
BNI
Applications
BNI
Applications
BNI
Applications
BNI
Applications
BNI
Latest promotions

Spend less time on DNA cleanup so you can do more science. The MSB Spin PCRapace is the fastest way to purify your DNA from PCR, restriction digestion, and...

New brilliant antibodies, and new lower prices!For flow cytometry reagents in general, \"bright is better.\" The violet-excitable BD Horizon™ BV421 and...

As an incentive to qualify our BSA, we are offering a 20% discount when you purchase your first 100g, 500g or 1000g of any grade of Bovine Serum Albumin....

It is not every day that you are given something for nothing. We are giving away additional spectrophotometer software.Cecil Instruments have enhanced the...

Did your supplier increase the price of Fetal Bovine Serum? Did they substitute the US Origin with USDA? Well say no more! Innovative Research is still...

We're so sure that you'll prefer Cayman Assay kits over your present brand that we're willing to give you a free assay kit to prove it!

For the past decade scientists have extensively used ATS secondary toxin conjugates to make their own targeted toxins for in vitro use.The ability to combine...

10% Discount on 2 Rabbit Polyclonal Antibody Service. With over 20 years experience, SDIX has developed into the premier US custom antibody producer,...

Bulk Cytokines with Custom Vialing.20 - 50% off cytokines, growth factors, chemokines and more...For a limited time Cell Sciences is offering substantial...

Are you planning to have a customised antibody made for your research?Since 2000, Everest has been producing a catalog containing thousands of affinity...
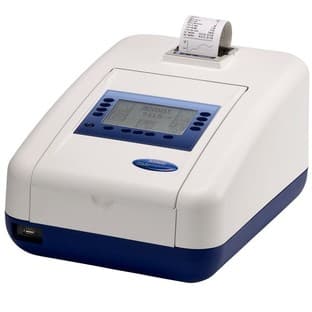
Jenway’s 73 series spectrophotometer range provides four models with a narrow spectral bandwidth of 5nm and an absorbance range of –0.3 to 2.5A,...
Top suppliers

United States Biological
230747 products
Carl Zeiss Microscopy
27 products
Promega Corporation
11 products
Panasonic Healthcare Company
5 products

Life Technologies
1 products

Nikon Instruments Europe
11 products

Olympus Europa Holding GmbH
3 products
Leica Microsystems, Inc.
10 products

GE Healthcare Life Sciences
2 products

Tecan Trading AG
19 products

Beckman Coulter, Inc.
1 products

AB SCIEX
3 products

BD (Becton, Dickinson and Company)
1 products
RANDOX TOXICOLOGY
5 products

Randox Food Diagnostics
6 products